Esistono una varietà di metodi che assegnano un punteggio locale a ciascun elemento di una sequenza (sia proteica che nucleotidica) a cui si assegna un preciso significato. Per esempio, nel paragrafo precedente il valore assegnato ad un determinato residuo amminoacidico, rappresenta la sua propensione locale (secondo l'intorno) ad essere più o meno in una fase apolare, mentre nel caso di sequenze nucleotidiche esistono metodi che assegnano a ciascuna base un punteggio che rappresenta la probabilità di essere all'interno di una zona codificante.
Un interessante problema che emerge dal fatto di avere sequenze
biologiche rappresentate da liste di numeri reali, è quello di
trovare la sottosequenza con la massima somma ([15]).
In modo più formale, se abbiamo una sequenza di numeri reali
![]() , il problema della massima sottosequenza
è quello di determinare la sottosequenza
, il problema della massima sottosequenza
è quello di determinare la sottosequenza
![]() tale per cui la somma degli elementi
tale per cui la somma degli elementi
![]() sia massima, ossia
sia massima, ossia
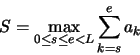
Un primo metodo che può essere utilizzato per la soluzione del
presente problema è l'algoritmo descritto in Figura ![[*]](crossref.png) .
Questo approccio conduce alla soluzione corretta, come si può
verificare eseguendo il codice, ma non è sicuramente la soluzione migliore.
Con maxsum3, si ha infatti che per ogni posizione della
sequenza (punto (1)), si riparte da capo a calcolare
dall'inizio (punto (2)) la somma (thisSum), che in realtà
viene valutata solo quando l'indice del secondo loop
.
Questo approccio conduce alla soluzione corretta, come si può
verificare eseguendo il codice, ma non è sicuramente la soluzione migliore.
Con maxsum3, si ha infatti che per ogni posizione della
sequenza (punto (1)), si riparte da capo a calcolare
dall'inizio (punto (2)) la somma (thisSum), che in realtà
viene valutata solo quando l'indice del secondo loop ![]() , diviene
maggiore o uguale all'indice corrente della sequenza (punto (3)).
, diviene
maggiore o uguale all'indice corrente della sequenza (punto (3)).
Possiamo comunque calcolare la complessità computazionale
dell'algoritmo esposto in Figura , ed è
dove con ![]() si é indicata la lunghezza della sequenza, (il calcolo si
esegue considerando la massima occupazione durante l'esecuzione),
mentre il tempo impiegato sarà
si é indicata la lunghezza della sequenza, (il calcolo si
esegue considerando la massima occupazione durante l'esecuzione),
mentre il tempo impiegato sarà
dove il tempo totale è calcolato come prodotto dei loops ((1),(2) e (3)) in quanto sono tutti e tre annidati.
Una maniera più "furba" per ottenere il medesimo risultato è mostrata
in Figura . Come si può notare un ciclo è stato
rimosso tenendo conto dell'inutilità evidenziata con maxsum3.
Con maxsum2 per ogni posizione ![]() della sequenza, si calcola
il nuovo segmento che puó ripartire da quella posizione e andare fino al
termine della sequenza. Analogamente a quanto fatto sopra si può verificare
che adesso
della sequenza, si calcola
il nuovo segmento che puó ripartire da quella posizione e andare fino al
termine della sequenza. Analogamente a quanto fatto sopra si può verificare
che adesso
mentre lo spazio occupato rimane il medesimo.
È possibile però fare ancora meglio! Esiste la possibilità infatti
di risolvere il problema con un algoritmo il cui tempo di esecuzione sia
lineare nella dimensione della sequenza (Figura ).
L'idea di base è la seguente:
siccome siamo interessati solo a valori la cui
somma siamo maggiore di ![]() , implica che ci basta tenere traccia
soltanto della migliore sottosequenza trovata da
, implica che ci basta tenere traccia
soltanto della migliore sottosequenza trovata da ![]() fino alla
posizione
fino alla
posizione ![]() della nostra sequenza, e considerare sottosequenze
nuove solo se la loro somma è
della nostra sequenza, e considerare sottosequenze
nuove solo se la loro somma è ![]() (#(2)).
In questo caso caso, possiamo confrontarla alla migliore trovata fino a questo
momento (#(3) ) e valutare se sia o meno il caso di tenere la nuova sequenza
come la mogliore ("best").
Se invece, la somma (o il valore attuale) è
(#(2)).
In questo caso caso, possiamo confrontarla alla migliore trovata fino a questo
momento (#(3) ) e valutare se sia o meno il caso di tenere la nuova sequenza
come la mogliore ("best").
Se invece, la somma (o il valore attuale) è ![]() , non interessa per nulla
continuare a calcolare la somma attuale, e si procede ripertendo da
, non interessa per nulla
continuare a calcolare la somma attuale, e si procede ripertendo da ![]() (#(2)).
(#(2)).
Come si può notare dal fatto che adesso esiste solo un ciclo (punto (1)), la complessità è
Questo paragrafo aveva lo scopo di mostrare l'affermazione fatta
all'inizio di questo capitolo, per cui dato un problema,
possono esistere numerose soluzioni non sempre
equivalenti. In casi reali la scelta di un algoritmo errato può rendere
il tempo di esecuzione inacettabile, e quindi trasformare il problema in
non risolubile.
Purtroppo esistono numerosi problemi, molti dei quali veramente rilevanti
nell'ambito della bioinformatica, che non hanno una soluzione polinomiale,
ossia il miglior algoritmo trovato richiede tempi di esecuzione esponenziali
o fattoriali nel numero dei dati (es. ![]() o
o ![]() ). Questi
problemi vengono trattati utilizzando algoritmi aprossimati o stocastici
(per una trattazione più dettagliata vedere [2], [4],
[7], [9], [10], [11]).
). Questi
problemi vengono trattati utilizzando algoritmi aprossimati o stocastici
(per una trattazione più dettagliata vedere [2], [4],
[7], [9], [10], [11]).
![\begin{figure}\small\begin{verbatim}def maxsum3(A=[]):
''' maxsum3(A[]) finds...
... Start=i
End=j
return([Start,End,maxSum])\end{verbatim}\normalsize\end{figure}](img108.png)
![\begin{figure}\small\begin{verbatim}def maxsum2(A=[]):
''' maxsum2(A[]) finds...
... Start=i
End=j
return([Start,End,maxSum])\end{verbatim}\normalsize\end{figure}](img112.png)
![\begin{figure}\small\begin{verbatim}def maxsum1(A=[]):
''' maxsum1(A[]) finds...
...he new best end
return([Start,End,maxSum])\end{verbatim}\normalsize\end{figure}](img119.png)